合作单位:Zhejiang University
参考文献:YZ Wu, SY Shen, JX Chen, et al. Metabolite asymmetric dimethylarginine (ADMA functions as a destabilization enhancer of SOX9 mediated by DDAH1 in osteoarthritis. Science Advances, 2023, 9(6): eade5584. DOI: 10.1126/sciadv.ade5584 (IF=13.6)
背景:
骨关节炎(OA)是最普遍的慢性关节疾病之一,影响多达三分之一的45岁以上人口。它主要是由老化和机械损伤引起的。骨关节炎的特征是软骨破坏、滑膜炎症、软骨下骨重塑和骨赘形成。富含糖胺聚糖的细胞外基质(ECM)是软骨的主要成分,由软骨细胞产生。在OA软骨细胞中,分解代谢调节因子释放多种炎症因子,包括白细胞介素-1β(IL-1β)、IL-6和肿瘤坏死因子-α(TNF-α),随后导致基质金属蛋白酶(MMP)和A分解素和具有血小板反应蛋白基序的金属蛋白酶(ADAMTS)的激活,最终导致ECM变性。软骨破裂加剧了关节中不平衡的机械应力,使OA发病机制陷入恶性循环。
在OA发病过程中,软骨细胞中的许多酶发生变化,导致一系列代谢变化,这使得某些代谢物有可能成为OA的诊断标志物。反过来,某些代谢途径的激活或抑制可能会加剧或阻止OA的进展。Corciul等证明,腺苷是三磷酸腺苷的代谢产物,在IL-1β的作用下减少,可以防止大鼠OA的发生。先前的一项研究表明,骨关节炎软骨细胞增加了胆固醇代谢。同时,胆固醇本身或关键胆固醇羟化酶CH25H和CYP7B1可通过RAR相关孤儿受体α(RORα)介导OA进展。
不对称二甲基精氨酸(NG,NG-二甲基-l-精氨酸,ADMA)是l-精氨酸的天然类似物。在精氨酸残基的翻译后甲基化过程中,它被一组称为蛋白精氨酸甲基转移酶和蛋白水解的甲基转移酶释放到细胞质中。肾脏负责清除循环系统中的游离ADMA。除肾脏排泄外,ADMA还可被二甲基精氨酸二甲氨基水解酶(DDAH)家族和丙氨酸-乙醛酸氨基转移酶2(AGXT2)水解。作为一种竞争性的内源性一氧化氮合酶(NOS)抑制剂,ADMA能够降低NOS活性,从而减少一氧化氮(NO)。该领域的研究主要集中在心血管疾病上。据报道,ADMA是高血压和充血性心力衰竭的主要独立危险因素,尤其是在肾功能衰竭患者中。此外,一些研究表明,ADMA与胰岛素抵抗,糖尿病和肝硬化有关。除NOS活性抑制外,ADMA能够通过促进辅助性T细胞1(TH1)–和TH17介导的免疫应答。在之前的研究表明,在肌肉骨骼系统中,ADMA升高会抑制骨形成。OA患者在滑液中的ADMA浓度通常高于血浆中的ADMA浓度。在代谢组学方面的数据显示,退化的软骨细胞中ADMA显著升高,具有最主要的统计学意义。因此,本文的目的是分析ADMA在OA中的作用以及它如何影响OA的进展。此外,还打算分析OA患者滑液中的ADMA是否可以作为OA的潜在诊断标志物
方案设计:
为了研究USP7与SOX-9之间的相互作用,探索了复合物之间的亲和力。通过与魔德科技技术团队沟通,根据客户提供的USP7蛋白和SOX-9蛋白信息,通过合理的蛋白对接,找到结合最稳定的蛋白-蛋白复合物结构。
主要结果:
作为一种内源性NOS抑制剂,ADMA可通过NO影响病理生理过程,尤其是在心血管疾病中。本文首先探讨了ADMA是否通过NOS/NO途径诱导软骨变性。DDAH1敲除或ADMA刺激不会改变NOS1和NOS3的蛋白表达,也不会激活软骨细胞或软骨中这些蛋白的磷酸化形式,而NOS2则不表达。出乎意料的是,DON在DDAH1缺失后没有表现出明显的变化,可能是因为它在软骨细胞的生理条件下浓度较低。TNF-α在静脉内皮细胞和成纤维细胞样滑膜细胞中的作用下ADMA升高。因此,应用TNF-α,并观察到软骨细胞中ADMA分泌增加,DDAH1过表达可以部分逆转。然而,在暴露于TNF-α或过表达DDAH1的软骨细胞中,NO没有显着改变。因此,ADMA诱导软骨细胞变性与NOS/NO途径无关。
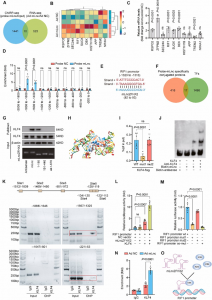
为了更好地了解其机制,研究了ADMA处理细胞的蛋白质组学。对差异表达的蛋白质进行亚细胞定位、基因本体和直系同源组簇分析。KEGG分析揭示了几种富集的OA相关途径,如ECM受体相互作用、TNF信号通路、细胞衰老和糖胺聚糖生物合成(图1A)。火山图显示分解代谢因子(MMP3和MMP13)、衰老标志物(P21)和炎症趋化因子(CCL2、CCL5和CXCL10)上调,合成代谢因子(聚集聚糖、COL2A1和SOX9; 图1B)。先前的一项研究表明,SOX9缺乏导致健康软骨中的蛋白多糖丢失,并导致创伤后使用OA后严重的软骨侵蚀SOX9FL/FL,ACAN-CreER系列T2航站楼小鼠。考虑到SOX9是蛋白质组学中变化最显著的蛋白质之一,也是软骨中最关键的转录因子之一,接下来重点关注SOX9是否是受ADMA影响的核心蛋白。RT-PCR显示,DDAH1的缺失不会显著改变SOX9 RNA水平(图1C)。然而,SOX9蛋白表达在暴露于ADMA或DDAH1缺失的软骨细胞中主要下调(图1D和E)。此外,免疫组化表明,关节注射PD 404182(图1F)或从DDAH1型FL/ FL,Col2-CreER型T2和DDAH1型FL/ FL,ACAN-CreER系列T2小鼠(图1G和H)。此外,SOX9在ADMA暴露细胞中的半衰期比对照细胞短,表明ADMA导致SOX9的不稳定性。MG132(一种蛋白酶体抑制剂)处理阻断了SOX9的衰变,而氯喹(一种溶酶体抑制剂)则没有,这表明SOX9降解依赖于蛋白酶体而不是溶酶体途径(图1I和J)。此外,ADMA诱导的SOX9衰变在过表达DDAH1的软骨细胞中被部分逆转(图1K)。值得注意的是,ADMA增加了泛素化SOX9的水平(图1L)。ADMA 共同通过泛素-蛋白酶体途径触发SOX9降解。
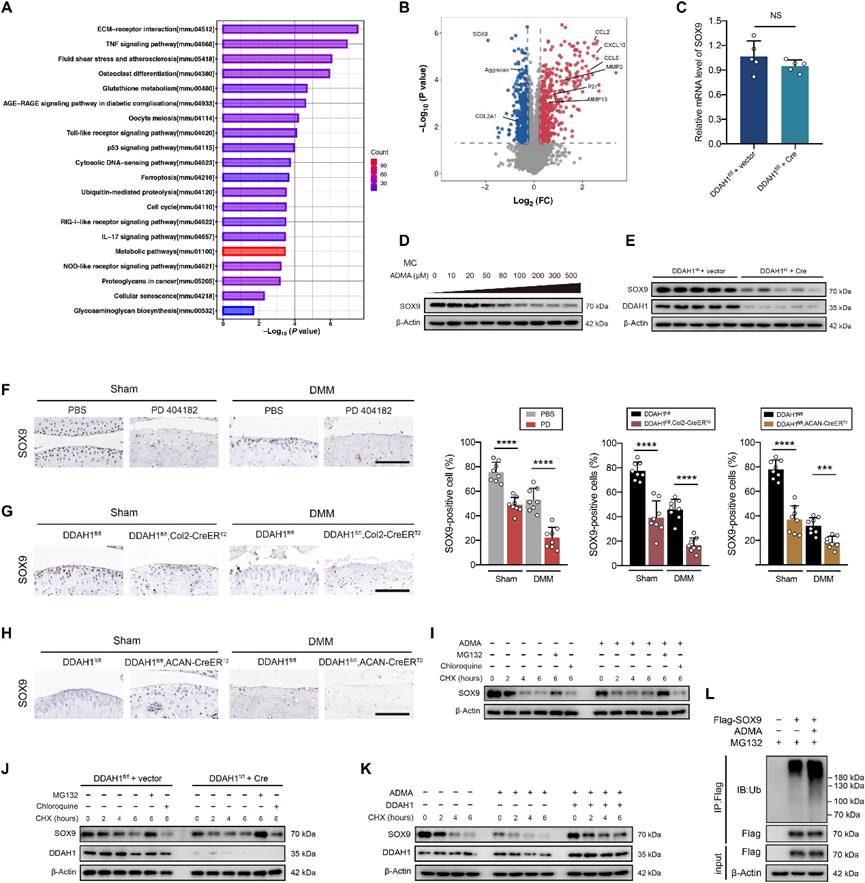
图1. ADMA通过泛素-蛋白酶体途径诱导SOX9降解
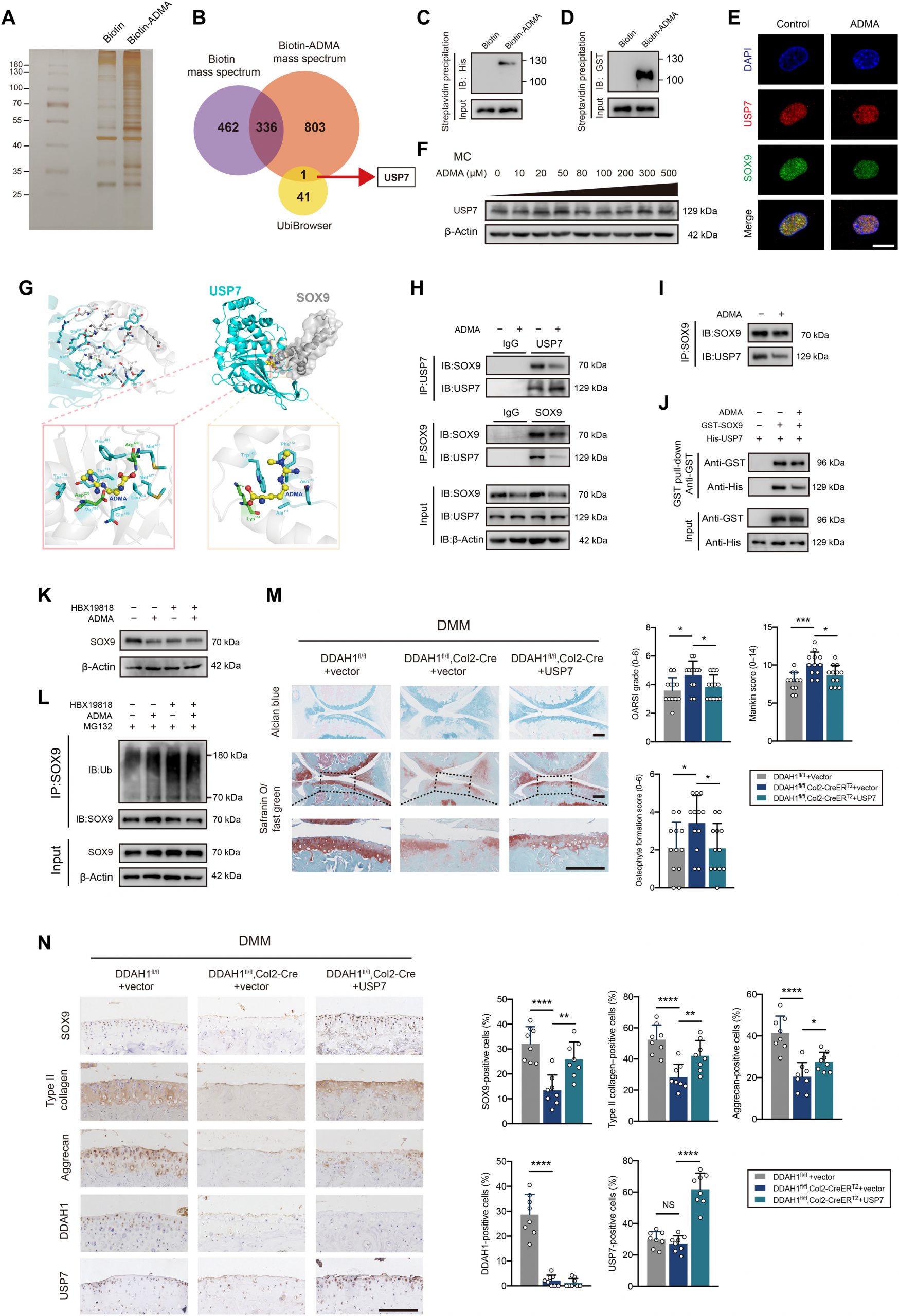
在证明ADMA通过泛素-蛋白酶体途径加速SOX9降解后,接下来旨在了解ADMA如何影响SOX9水平。通过合成了生物素-ADMA,并将其与软骨细胞裂解物一起孵育。银染与MS一起表明,生物素-ADMA可以与一系列蛋白质结合。通过重叠了与UbiBrowser数据库预测的生物素ADMA和SOX9相关E3泛素连接酶或去泛素化酶(DUB)特异性相互作用的蛋白质,发现DUB USP7可能参与其中(图2A和B)。之前的一项研究报告称,ATDC5细胞中的USP7敲低会降低SOX9表达。HBX19818(一种特异性的USP7抑制剂)降低了软骨细胞中的SOX9蛋白水平。免疫沉淀结果显示,USP7能够还原泛素化的SOX9。USP7和ADMA之间的相互作用通过蛋白质印迹法验证。出乎意料的是,ADMA在质谱数据中与SOX9相互作用,这进一步通过蛋白质印迹法得到证实。为了探究ADMA是否直接与这两种蛋白结合,应用谷胱甘肽S-转移酶(GST)-SOX9重组蛋白和His-USP7重组蛋白,并将它们与生物素-ADMA分别孵育,发现SOX9和ADMA(或USP7和ADMA; 图2C和D)。共聚焦显微镜显示USP7和SOX9共定位。用ADMA处理细胞后,SOX9的荧光信号减弱,而USP7没有改变(图2E),这通过蛋白质印迹法验证(图2F)。
应用分子对接预测ADMA、USP7和SOX9之间的相互作用模型(图2G)。USP7和SOX9共有12个氢键,这导致这两个蛋白之间有很强的相互作用。一方面,ADMA与由Tyr224、Asp295、Val296、Gln405、Leu406、Met407、Arg408、Phe409、Met410和Tyr514组成的USP7的空腔进行空间结合,ADMA中的氮原子和氧原子形成两个氢键:Asp295和Arg408。同时,ADMA还在USP7空腔周围与Tyr224、Val296、Gln405、Leu406、Met407、Phe409、Met410、Tyr514等形成强疏水相互作用。另一方面,ADMA与Asn110、Ala111、Phe112、Trp143和Lys151形成的SOX9的螺旋角空腔在立体上完美对接,ADMA的氧原子与SOX9的Lys151形成氢键。同时,ADMA与SOX9空腔周围的Asn110、Ala111、Phe112、Trp143之间也存在较强的疏水相互作用。因此,ADMA能够结合到SOX9和USP7之间的相互作用区域,主要阻断它们的结合。共免疫沉淀证实了SOX9和USP7之间的相互作用,这种相互作用可以被ADMA削弱(图2H)。考虑到ADMA刺激导致免疫印迹阶梯中SOX9蛋白水平不平衡,仔细调整SOX9蛋白水平以保证等效的SOX9初始水平,但仍发现ADMA处理的软骨细胞中USP7和SOX9之间的相互作用减少(图2I)。GST下拉实验证实了USP7和SOX9之间的直接结合,ADMA可以部分阻断这种结合(图2J)。
为了进一步验证假设,在ADMA处理细胞之前使用HBX19818。在抑制USP7后,ADMA未能影响SOX9的表达和SOX9粘附的泛素(图2K和L)。此外,在DMM手术后1周,将USP7AAV关节注射到DDAH1fl/fl、Col2-CreERT2小鼠的膝关节。值得注意的是,通过免疫组化检测,软骨损伤在DMM手术后8周得到了显著修复,SOX9、II型胶原和聚集蛋白的表达增加(图2M和N)。此外,检测了OA患者接受全膝关节置换术后胫骨平台配对损伤和未损伤软骨中SOX9和USP7的蛋白表达。软骨组织中SOX9和USP7蛋白表达没有明显的相关性,这在一定程度上与本文的发现一致,即USP7酶活性而不是其表达参与OA的进展。总之,这些数据支持ADMA阻断USP7对SOX9的去泛素化作用的观点。
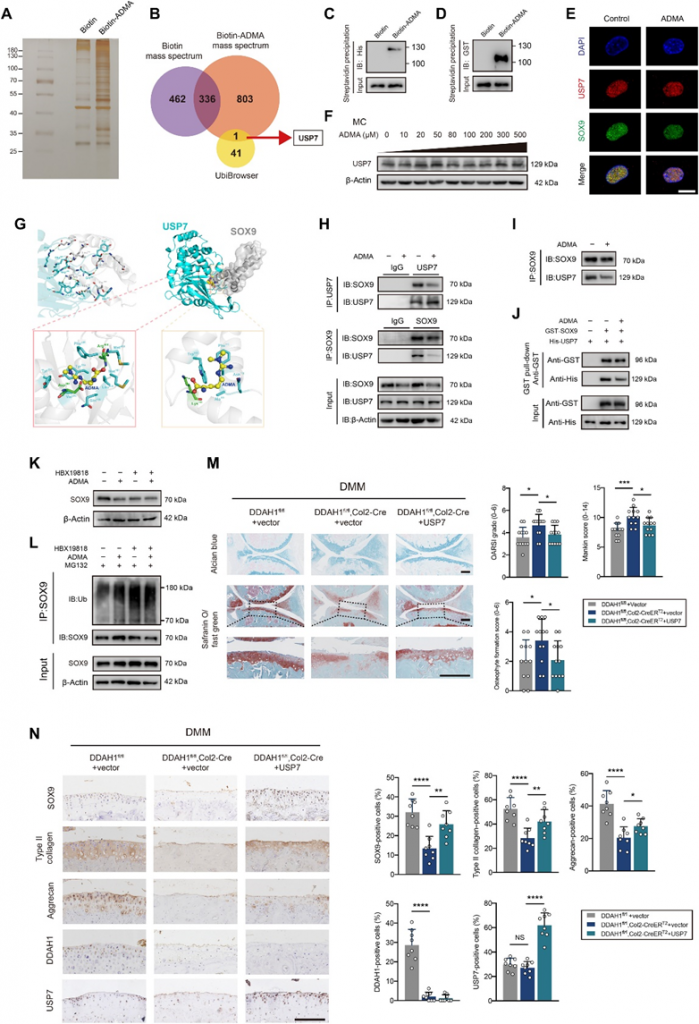
图2. ADMA阻断SOX9和USP7之间的相互作用
结论:
骨关节炎是一种退行性疾病,伴有一系列代谢变化和许多酶的改变。在这里,本文报道了二甲基精氨酸二甲氨基水解酶-1(DDAH1)的下调伴随着变性软骨细胞和OA样本中不对称二甲基精氨酸(ADMA)的增加。ADMA水解酶DDAH1的整体或软骨细胞条件敲除加速了小鼠OA的发展。ADMA诱导软骨细胞变性和衰老,减少细胞外基质沉积,从而加速OA的进展。ADMA同时与SOX9及其去泛素化酶USP7结合,阻断USP7对SOX9的去泛素化作用,从而导致SOX9降解。OA患者滑液中ADMA水平升高,对OA诊断具有良好的敏感性和特异性的预测价值。因此,激活DDAH1以降低ADMA水平可能是OA治疗的潜在治疗策略。