生物数据格式
一、生物数据
在生物信息学领域,对生物数据进行处理和分析往往是研究生物学问题的起点。从技术上讲,已经有多种数据存储的解决方案,大量生物数据随着人类基因组测序的完成被研究挖掘出来。然而需要注意的是,由于每个人的研究角度和方法不同,造成他们对数据整合的理解和解释有差异。数据存储的多样性和数据解释的多样性两者共同为研究者的数据检索带来不便,另外,高通量筛选在近年来的快速发展也促成生物数据量的迅猛增长。建立和使用可重复和可扩展的生物数据存储方式尤为重要。
生物数据格式表示被存储的生物信息,是我们通过文件表示生物信息的一种具体方式。研究者通过下载相应的文件获取写入文件中的生物信息。在历史上出现过不同格式的数据表示相同的信息,一般的,生物数据格式应该包含生物本身的信息(如蛋白质序列组成)和元数据——额外的描述信息(如蛋白质名称)。区分这两部分的工作催生出了大量不同的生物数据格式。获取数据的最终目的是分析,处理和传输它们的便利性也是研究者关注的重点。目前,生物信息学研究中常用的生物数据格式有FASTA格式、GenBank格式、EMBL格式等,这里对这些数据格式的组成以及简单处理进行介绍。
1. FASTA格式
FASTA(fast alignment)格式是一种基于文本的格式,源于一款问世于20世纪80年代的软件FASTP。它采用单字母编码表示氨基酸或核苷酸,主要由两部分组成,即元数据信息和序列信息本身,通常文本的第一行以“>”开头,“>”后则是标识符,此行称为声明行或定义行。蛋白质的序列信息以氨基酸的单字母形式排列于前者的下方。由于格式的简单,绝大多数蛋白质序列搜索平台都以它为数据的输出格式。
对于FASTA文件的获取,如前所说可以通过很多平台,不同平台的操作方式可能有不同,下面以蛋白质数据库(PDB)为例进行介绍。首先登录蛋白质数据库网站(见“蛋白质数据库”一节),在如图所示的搜索框中键入蛋白质的PDB ID(如1CP9),如图所示;在随后弹出的页面中点击Download Files按钮,选择FASTA Sequence,蛋白质序列的FASTA格式文件开始下载;下载完成后用文本编辑软件打开即可。
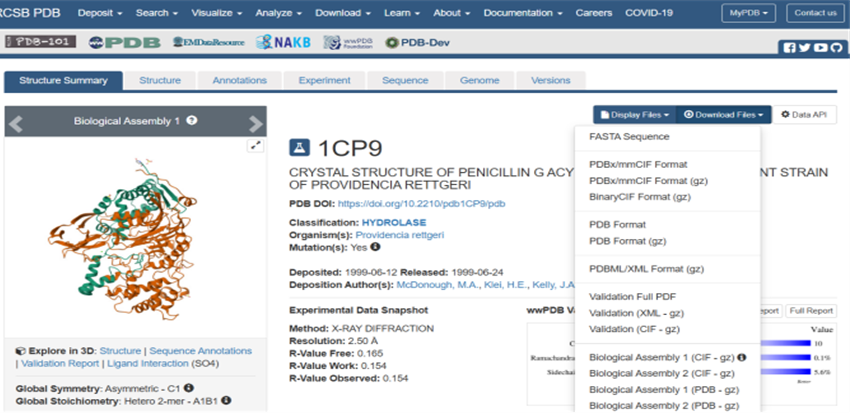
图1 点击Download Files按钮,选择FASTA Sequence
数据的分析过程不可避免地要经过数据传输,当前各大数据库中的数据量呈指数级增长,传输之前对文件进行压缩不但有利于缩减单个文件所占的存储空间,还能提高数据传输速度。对FASTA格式文件的压缩方法主要有DELIMINATE、BIND计算、和MFCompress方法,它们都是缩减字符或对字符进行编码,最后使用一些如7-Zip的压缩器对处理后的文件进行压缩。以压缩核苷酸序列文件为例,DELIMINATE将标识码和序列数据分开处理。分两个阶段压缩序列数据,非必要的字符等信息在第一阶段被记录并删除,所得的文件在第二阶段通过重新编码的方式被处理,在此过程中生成的各种文件由7-Zip压缩器压缩为最终的文件。与其他压缩方法相比,DELIMINATE耗时更短;并且现在大多数工作站都具备两个内核,所以DELIMINATE压缩的两个步骤可以同时进行,效率更高。
- GenBank格式
GenBank格式主要应用于DNA序列,它是美国国家生物技术信息中心(National Center for Biotechnology Information, NCBI)所建立的DNA序列数据库使用的数据格式。这种数据格式处理起来比较简单,可以满足高通量测序技术高速发展而带来的数据存储需求。
对基因序列等生物信息的研究与相关已有的研究文献密不可分,基因记录与已发表文献的联系是当代生物医学研究中的重要组成部分。记录基因数据的出版物需要同一些如PubMed的文献数据库建立链接,保证研究者可以获得更多实验详细信息以指导实践。在PubMed收录的期刊中,更多的基因序列文献可以在Plos Biology中找到。Wang等人在一篇研究蝗虫基因组的文章中,通过将特定的字符串插入文章的在线版本,点击这行字符读者就可以进入DNA数据库完整地看到GenBank格式的蝗虫基因组,实现了比较完美的信息与文献之间的链接。


