乳腺癌治疗新途径——CD47-SIRPα阻断诱导铁下垂激活
合作单位:Shandong University
参考文献:ZJ He, H Zhou, Y Zhang, et al. Oxygen-boosted biomimetic nanoplatform for synergetic phototherapy/ ferroptosis activation and reversal of immune-suppressed tumor microenvironment. Biomaterials, 2022, 290: 121832. DOI: 10.1016/j.biomaterials.2022.121832 (IF=14.0)
背景:
乳腺癌在世界卫生组织2021年发布的报告中被认定为最常见的恶性肿瘤类型,严重威胁着女性的身心健康。在临床上,手术、辅助化疗和放疗是主要的治疗方式,但其疗效仍不充分。光动力治疗(PDT)作为一种无创、有效、时空可控的抗癌方式,在肿瘤联合治疗中受到广泛关注。PDT利用局部氧气(O2)、光敏剂和特定光源产生具有高毒性的活性氧(ROS),引发肿瘤细胞凋亡死亡。最近的大量研究也表明,PDT还可能增强肿瘤的免疫原性,增强机体的抗肿瘤免疫。
然而,为了获得满意的PDT效益,仍然存在几个重要的问题。首先,新出现的证据支持这样的观点:肿瘤细胞对凋亡既有固有的抗性,也有获得性的抗性,这增加了PDT治疗失败和治疗后复发的风险。因此,需要将其他细胞死亡模式与PDT相结合。Ferroptosis是一种不同于凋亡、坏死和自噬的铁依赖性细胞死亡模式,由Stockwell等于2012年首次提出。简单地说,羟基自由基(OH)可以通过肿瘤细胞中亚铁离子(Fe2+)和过氧化氢(H2O2)的芬顿反应产生,将多不饱和脂肪酸(PUFA)氧化为脂质过氧化物(LPO)。LPO可破坏细胞的结构和完整性,引起铁下垂。通常,铁下垂可通过经典和非经典模式触发。经典诱导模式下,通过干扰膜保护触发铁下垂;谷胱甘肽过氧化物酶4(GPX4)作为细胞抗铁死亡的主要保护剂,在许多研究中一直被认为是主要靶点。在非经典模式下,细胞内Fe2+过载诱导铁下垂,导致Fenton反应增强,产生充足的OH,最终触发铁下垂。目前,有多种策略可以单独或联合调节上述两种模式,以更有效地诱导铁下垂。例如,作为一种典型的刺激剂,erastin通过调节系统Xc–-谷胱甘肽(GSH)/GPX4轴来激活ferroptosis。以RAS选择性致死小分子3(RAS-selective lethal small molecule 3, RSL3)为代表的GPX4抑制剂可直接灭活GPX4,引发铁下垂。此外,结合其他策略,RSL3对铁下垂的激活作用得到增强。如Guo等人设计了含有硝基咪唑基团的RSL3负载的铁死亡胶束,可同时消耗NADPH、GSH和硫氧还蛋白,从而强力触发4T1细胞的铁死亡。为了实现细胞内Fe2+的过载,hemin被用来上调血红素氧化酶-1(HMOX-1)的表达,从而诱导血红素降解和Fe2+释放。从另一个角度来看,Zhu等人精心开发了可将近红外光(NIR)转换为紫外线(UV)的上转换纳米颗粒;Fe3+在近红外照射下会还原为Fe2+。
此外,新兴的研究报道了PDT和铁下垂激活的结合,以获得提高的抗癌效率。例如,Zhu等人利用Erastin激活ferroptosis,增强Ce6的PDT效率。Meng等人通过GSH消耗方式诱导铁下垂成功增强了PDT。Li等人制备了负载Ce6和丁硫氨酸亚砜胺的介孔包被上转换纳米颗粒,也可以放大铁致死亡和凋亡细胞死亡。有趣的是,除了通过激活铁死亡来增强PDT外,最近的研究表明,PDT还可能通过消耗GSH和刺激干扰素-γ (IFN-γ)的分泌来促进铁死亡,干扰素-γ会下调系统Xc–的表达。考虑到铁下垂激活与PDT之间的串扰的复杂性,可能存在其他协同机制;迫切需要全面讨论它们的相互作用,以实现它们的有效合作。
其次,TME中的缺氧阻碍了PDT的ROS生成,这是TME的固有特征,并且在消耗O2的PDT过程中会进一步加剧。此外,缺氧还可能使肿瘤相关巨噬细胞从免疫增强型M1型向免疫抑制性M2型转变,促进肿瘤细胞的免疫逃逸。因此,缓解PDT过程中肿瘤缺氧具有重要意义。
第三,尽管有报道称PDT通过诱导免疫原性细胞死亡(immunogenic cell death, ICD)激活T细胞免疫,但其效率远远不够,这可能与TME中存在多种免疫逃避机制有关。因此,将PDT与免疫检查点阻断(immune checkpoint blockade, ICB)联合治疗一直是一种常见的做法,大多数研究都集中在PD1/PDL1阻断上。然而,并非所有患者都能对PD1/PDL1阻断产生反应,这提醒我们要关注其他ICB机制,为患者提供更多的机会。
巨噬细胞作为先天免疫系统不可缺少的组成部分,在实体肿瘤中大量存在,在肿瘤的防御中起着至关重要的作用。然而,CD47(肿瘤细胞表面)与信号调节蛋白α (SIRPα)(巨噬细胞表面)的结合向巨噬细胞传递了“不要吃我”的信号,从而避免了巨噬细胞对肿瘤细胞的清除。为了阻断CD47- sirp α的相互作用,多种CD47阻滞剂已经在临床试验中被开发出来。由于CD47也广泛分布于正常细胞(尤其是红细胞)表面,阻断CD47- sirp α可能引起贫血、血小板减少等副作用;因此,如何实现CD47阻滞剂的肿瘤特异性递送成为一个关键问题。与目前广泛研究的CD47单克隆抗体相比,CD47抑制肽更适合于纳米载体修饰和递送。PEP20是一种具有代表性的CD47抑制肽,其有效性和安全性已被体外和体内实验证明。
然而,PEP20以自由形式给药,可能缺乏肿瘤靶向能力,在体内被快速清除;因此,需要合理设计纳米载体来精确递送PEP20。为了突破上述PDT的研究瓶颈,该工作设计了一种新的纳米平台,整合了O2自供PDT、铁凋亡激活和双免疫激活(T细胞和巨噬细胞免疫)的功能。如图1A所示,牛血清白蛋白(BSA)被用作Ce6和血红素(H)的载体,形成Ce6/血红素共载的牛血清白蛋白纳米颗粒(CH/BSA NP)。具有肿瘤亲和性的M1巨噬细胞膜被PEP20修饰,形成PEP20修饰膜(MP),用于伪装CH/BSA NP,得到MP@CH/BSA NP。采用基质金属蛋白酶-2 (matrix metalloproteinase-2, MMP-2)敏感肽序列(-PLGPAG-, L)作为PEP20与膜之间的连接物,设计由TME中过表达的MMP-2进行切割(方案1A);释放的PEP20有望阻断TME中CD47-SIRPα的相互作用。
MP@CH/BSA NP强烈诱导4T1细胞凋亡和铁下垂,这是体外和体内数据的结果。hemin参与MP@CH/BSA NP制剂通过其过氧化氢酶(CAT)模拟活性缓解肿瘤缺氧,从而如预期的那样改善Ce6的PDT效率。关于铁下垂,MP@CH/BSA NP被观察到通过经典(解除GPX4途径)和非经典(诱导Fe2+过载)模式激活铁下垂。有趣的是,除了hemin在消耗GSH和上调HMOX1表达中的作用外,Ce6还首次在经典和非经典模式下增强了铁凋亡的激活。具体来说,Ce6通过以下途径增强铁凋亡:(A) Ce6被证明在激光照射下触发MP@CH/BSA NP溶酶体逃逸;体外释放结果也证实了其能加速MP@CH/BSA NP中血红蛋白的释放。Ce6共同促进细胞质中血红素的释放,从而促进血红素诱导的铁下垂激活。(B)由于溶酶体是细胞内Fe2+的储库,在MP@CH/BSA NP的溶酶体逃逸过程中,突然释放的Fe2+加重了细胞内Fe2+过载,通过非经典模式激活铁下沉。(C)根据先前的研究,Ce6介导的PDT消耗细胞内GSH并下调肿瘤细胞的系统Xc–表达,这些共同促进了铁凋亡的经典激活。
在免疫调节作用方面,MP@CH/BSA NP同时增强巨噬细胞和T细胞免疫,全面逆转免疫抑制的TME,从而消除原发肿瘤,抑制肿瘤转移。在巨噬细胞免疫方面,研究创新设计了MMP2响应的纳米囊泡,用于CD47抑制肽(PEP20)的递送,以实现ICB目的。MP@CH/BSA NP在体外和体内均能很好地保留PEP20的生物活性,并显著增强巨噬细胞介导的4T1细胞吞噬作用。有趣的是,首次发现PEP20可以通过刺激IFN-γ分泌和下调系统Xc–来致敏铁下垂,创造性地连接CD47-SIRPα阻断和铁下垂治疗,为肿瘤联合治疗提供更多可能性。
综上所述,MP@CH/BSA NP通过简单的步骤在温和的条件下制备,并首次集成了O2增强的PDT,铁凋亡激活和双免疫激活,为解决上述阻碍PDT的问题提供了策略。研究揭示了PDT-铁下垂激活相互作用的机制,为两种治疗方式的结合奠定了理论基础。此外,新发现的CD47-SIRPα阻断诱导的铁下垂激活可能为乳腺癌的联合治疗提供新的途径。
方案设计:
为了研究牛血清蛋白与血红素、二氢卟吩化合物的相互激活机制。通过与魔德科技技术团队沟通,拟采用分子对接和分子动力学模拟方法研究了牛血清蛋白与血红素、二氢卟吩化合物的分子自组装过程。
主要结果:
MP@CH/BSA NP的制备和表征
如图1A所示,MP@CH/BSA NP的制备过程分为三个步骤:(1)CH/BSA NP的自组装,(2)巨噬细胞极化、膜分离和修饰,(3)膜伪装。在步骤1中,通过自组装工艺在超声下获得CH/BSA NP。根据UV-vis-NIR扫描数据,C和H特征峰的存在证实了C和H的成功加载(图1E)。
在CH/BSA NP制备过程中,观察超声时间和药载比对CH/BSA NP的大小和载药效率(DLE)的影响;确定最佳超声时间为4 min,药载比为3:8。通过改变C与H的投料比为1:1 ~ 3:1 (w:w),制备出三种CH/BSA NP配方,即CH/BSA NP 1、2和3,其粒径相似,但C与H的负载比不同。由于根据细胞毒性结果选择CH/BSA NP 2为最佳配方,除非另有说明,否则在后续实验中使用CH/BSA NP 2。所得CH/BSA NP水化直径为123 nm(图1C),粒径分布较窄(图1B)。TEM图像显示CH/ BSA NP呈球形(图1D-a)。然而,CH/BSA NP表现出不理想的稳定性,在PBS中孵育或不孵育FBS(10%)后,其直径和多分散性指数(PDI)大大增加(图1F-a, b)。
为了解决这一问题,采用了膜伪装策略,该策略已被证明可以增强内NP的稳定性。由于巨噬细胞膜伪装也可以避免单核吞噬细胞系统(MPS)对NP的清除,因此将CH/BSA NP包被巨噬细胞膜。此外,考虑到M1巨噬细胞对非极化巨噬细胞(M0巨噬细胞)具有优越的肿瘤靶向能力,采用脂多糖(LPS)刺激和膜分离后获得的M1巨噬细胞膜覆盖NP。通过表达iNOS (M1生物标志物)证实了膜极化的成功(图1H-a)。接下来,在膜巯基化(巯基含量从0.0164增加到0.3844 μmol mL-1,为化学修饰留下足够的反应位点)和巯基点击化学之后,通过MMP-2敏感连接剂L(肽序列:- PLGLAG -)将PEP20偶联到M1膜上,得到MP(图1A:步骤2)。最后,在步骤3(图1A),采用有充分记录的挤压方法制备MP包被的NP (MP@CH/BSA NP)。
从TEM图像中可以直观观察到,与裸CH/BSA NP相比,MP@CH/BSA NP呈现出明显的“核壳”结构,表明CH/BSA NP被膜薄壳伪装,膜薄壳厚度约为~10 nm(图1D-b)。在DLS分析中,由于膜伪装,粒径从122.97 nm (CH/BSA NP)增加到137.33 nm (MP@CH/BSA NP)(图1C)。zeta电位从-3.42 mV (CH/BSA NP)变化到-10.17 mV (MP@CH/BSA NP)(图1C),接近膜电位(MP)。此外,通过SDS-PAGE和Western blot也可以验证MP伪装的成功和功能蛋白的良好保存(图1G和H-b)。
为了进一步证明PEP20-L-Mal在M@CH/BSA NP上的保存并计算共轭效率(CE),使用荧光素标记的PEP20-L-Mal修饰M并产生MPFITC。计算出FITCP在M@CH/BSA NP表面的CE为21.77%。
综上所述,MP@CH/BSA NP可以按照图1A所示的三个步骤轻松制备。所得MP@CH/BSA NP呈核壳结构的球形(图1D),平均直径为137.33 nm, zeta电位为10.17 mV(图1C)。与裸NP相比,MP@CH/BSA NP的大小和PDI在有/没有FBS的PBS中几乎没有变化,这表明伪装策略有效地增强了NP的稳定性(图1F-a, b和表S2, S3)。此外,即使载体浓度高达1 mg mL-1,溶血率也保持在1%以下,这表明我们的纳米载体具有理想的生物安全性。
为了研究MP@CH/ BSA NP装载货物的释放行为,采用透析法。如图1I所示,MP@CH/BSA NP对H和C的释放呈持续模式。有趣的是,观察到激光照射可以加速药物释放(图1I);这一现象可能是由于Ce6产生了大量的ROS,破坏了MP@CH/BSA NP的外膜,从而消除了药物释放的屏障。这种激光加速药物释放谱可以通过施加局部照射来加速药物在目标部位的释放,这可能有助于避免循环中的不成熟释放,并保证在肿瘤部位充分释放。
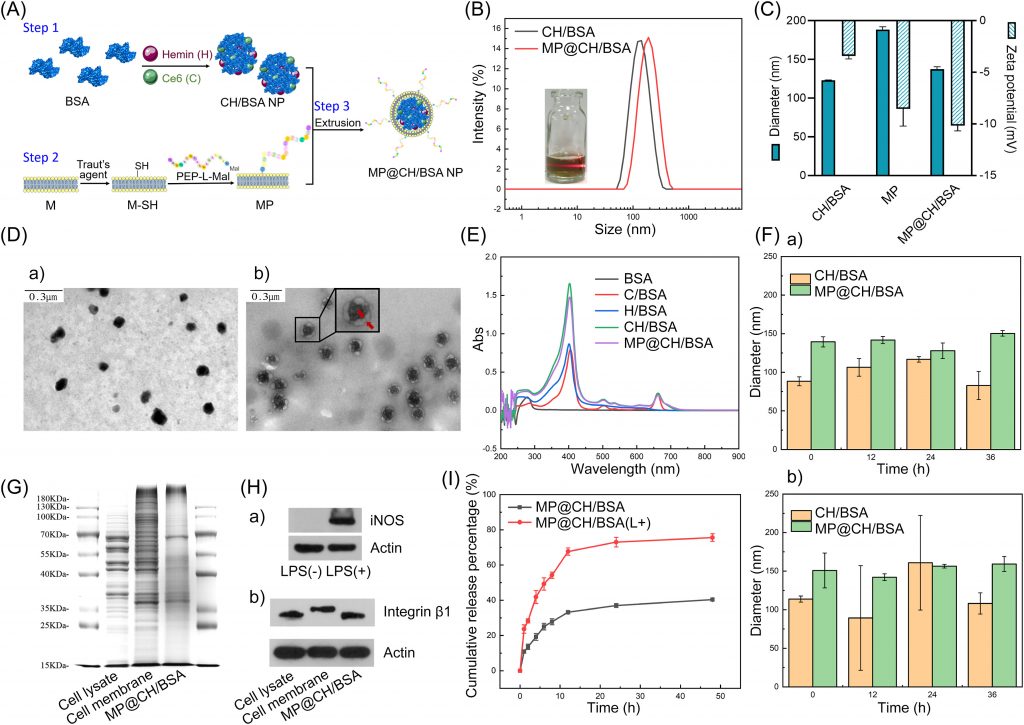
图1 MP@CH/BSA NP的制备及理化性质研究。(A) MP@CH/BSA NP的制备过程。(B) CH/BSA和MP@CH/BSA NP的粒径分布及MP@CH/BSA NP溶液外观。(C) CH/BSA NP、MP和MP@CH/BSA NP的直径和ζ电位(n = 3)。(D) CH/BSA (a)和MP@CH/BSA NP (b)的TEM图像。比尺:0.3 μm。(E)不同配方的紫外-可见吸收光谱。(F) 37◦C PBS (a)和10% FBS/PBS (b)孵育36 h后CH/BSA和MP@CH/BSA NP的粒径变化(n = 3)。(G)巨噬细胞裂解液、巨噬细胞膜和MP@CH/BSA NP中蛋白质组成的SDS-PAGE分析。(H) Western blot分析LPS处理前后巨噬细胞膜中iNOS和巨噬细胞裂解液、膜和MP@CH/BSA NP中整合素β1的表达水平。(I)激光触发MP@CH/BSA NP在PBS (pH 7.4) + 0.5% Tween 80中释放血红蛋白(n = 3)。
CH/BSA NP的自组装机理
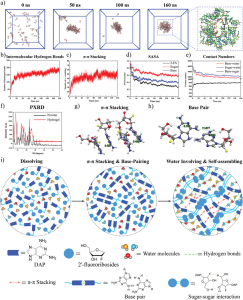
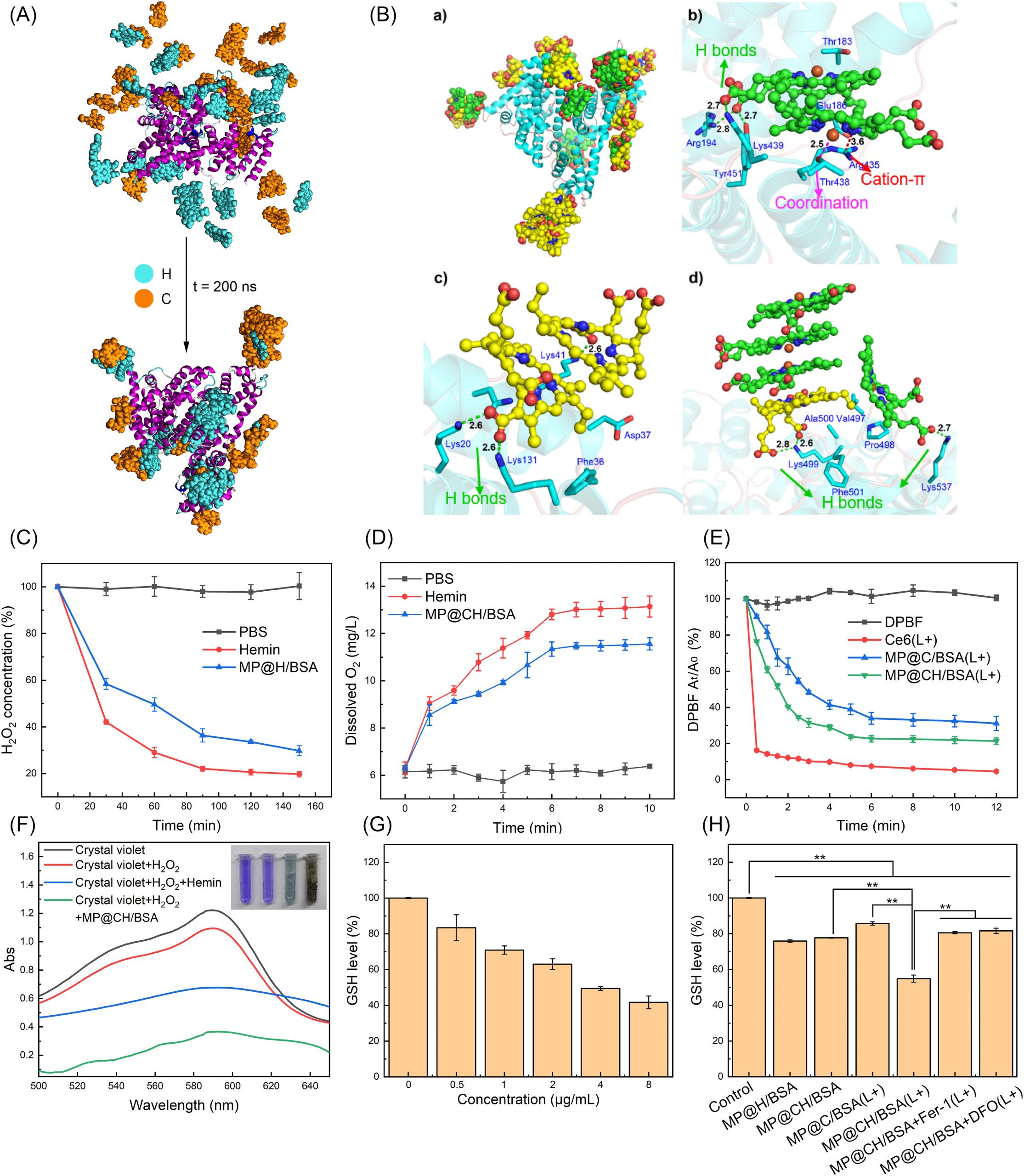
如前一节所述,CH/BSA NP可以通过三种组分在水溶液中的自组装过程获得。为了探索这种自组装过程并了解分子间结合的驱动力,进行了分子动力学(MD)模拟。图2A显示了MD模拟前后CH/BSA NP的构象。最初,H和C随机分布在BSA周围;MD模拟200ns后,大部分H和C聚集结合在BSA蛋白表面,形成更稳定的组装结构。通过分析MD模拟过程中均方根偏差(RMSD)和旋转半径(Rg)的变化发现,模拟100 ns后,体系的这两个值趋于稳定,说明BSA蛋白与H和C分子结合后结构达到稳定状态。通过分析均方根波动(RMSF)的变化可以看出,模拟过程中BSA蛋白的整体柔韧性较小,大部分RMSF值都在0.2以下,说明模拟过程中BSA处于相对稳定的状态。在MD模拟过程中,H或C与BSA之间的氢键逐渐增强,在100 ns后分子间氢键数基本保持稳定,其平均值分别为8.00和9.00。此外,溶剂可及表面积(SASA)分析结果显示,CH/BSA在模拟过程中可以形成相对致密的纳米颗粒结构,SASA值降低了29.69%。
为了分析H、C与牛血清白蛋白的分子识别机制,研究了H、C与牛血清白蛋白的结合模式。如图2B-a所示,H和C可以聚集在BSA表面的不同区域。选取三个具有代表性的结合区进行相互作用分析;如图2B-b、c、d所示,H、c主要通过氢键相互作用与BSA蛋白结合。具体来说,参与氢键的氨基酸有Lys20、Lys41、Lys131、Arg194、Lys439、Tyr451、Lys499和Lys537,也就是说,主要是带正电的赖氨酸和精氨酸,它们可以与H和C结构中的羧基形成氢键和静电相互作用。此外,H结构的离子也可以与BSA中的氨基酸形成配位,如Thr438。此外,H和C均含有芳香环结构,可与牛血清白蛋白氨基酸(如Arg435)相互作用形成阳离子-π相互作用。综上所述,氢键、配位和阳离子-π相互作用共同驱动了CH/BSA NP的形成。
图2 CH/BSA NP (A-B)的自组装机理及MP@CH/BSA NP (C-H)的催化氧、ROS和OH生成及GSH消耗评价(A) CH/BSA NP在水溶液中的自组装过程。(B) BSA蛋白C、H与氨基酸残基的结合模式。a) MD模拟得到的CH/ BSA NP结构。b-d)三个具有代表性的结合区域(绿色和黄色球棒模型代表H和C分子,青色棒模型代表BSA中的氨基酸残基;绿色、紫色和红色虚线分别代表氢键、配位和阳离子-π函数)。
(C) PBS、hemin溶液和MP@H/BSA溶液中H2O2浓度的变化(n = 3)。(D) PBS、hemin溶液和MP@CH/BSA溶液中溶解O2浓度的变化(n = 3)。(E)以DPBF为探针激光照射后游离Ce6、MP@C/BSA NP和MP@CH/BSA NP的ROS生成(n = 3)。(F)不同处理后结晶紫降解的紫外-可见吸收光谱。(G) MP@CH/BSA NP在不同浓度下的谷胱甘肽消耗(n = 3)。(H)不同配方的谷胱甘肽消耗(n = 3)。
MP@CH/BSA NP的CAT模拟活性、ROS生成和GSH耗竭
MP@CH/BSA NP由于hemin的存在,预计会表现出模仿cat的活性。为了证实这一点,记录了PBS、hemin溶液和MP@CH/BSA NP溶液中O2和H2O2浓度在孵育期间的变化,结果表明MP@CH/BSA NP可以分解H2O2并产生O2(图2C和D)。接下来,以DPBF作为ROS探针,评估MP@CH/BSA NP在水溶液中的PDT电位。如图2E所示,与不含血红素的MP@C/BSA NP相比,MP@CH/BSA NP确实表现出增强的ROS生成能力,这可能归因于其模拟cat的活性。
为了初步评价MP@CH/BSA NP的吊铁活化电位,采用紫外-可见分光光度计检测芬顿反应生成的⋅OH,并记录不同处理后的结晶紫吸收光谱。如图2F所示,经过H2O2处理后,结晶紫的吸收强度可以忽略不计,说明OH生成可以忽略不计。与此相反,经hemin + H2O2处理后,晶体紫荧光明显减弱,这表明了OH的产生,这可能与hemin介导的Fenton反应有关。令人鼓舞的是,与hemin相比,MP@CH/BSA NP + H2O2能产生更多的OH,这表明MP@CH/BSA NP在铁下垂激活中的潜力更大。
此外,MP@CH/BSA NP在水溶液中表现出消耗谷胱甘肽的能力,并且随着MP@CH/BSA NP浓度的增加,谷胱甘肽的消耗量也随之增加(图2G)。为了了解MP@CH/BSA NP中不同成分对GSH消耗的贡献,记录了含有不同配方的溶液中GSH水平的变化。如图2H所示,与对照组相比,各制剂均能有效消耗GSH (p < 0.01),这表明纳米平台中的Ce6和hemin都能消耗GSH,这也是之前研究报道的。与MP@C/BSA NP (L+)和MP@CH/BSA NP相比,MP@CH/BSA NP (L+)对GSH的消耗作用最强,进一步证明了Ce6和hemin在强化GSH消耗中的协同作用。
结论:
提出了一个新的仿生纳米平台(MP@CH/ BSA NP),其中整合了O2增强的PDT,铁上吊诱导(通过经典和非经典模式)和CD47-SIRPα阻断活性,在体外和体内均表现出良好的抗癌活性。有趣的是,这三种治疗方式之间存在多重促进作用。例如,Ce6介导的PDT被创新地验证可以通过经典(下调GPX4途径)和非经典(诱导Fe2+过载)模式激活铁上吊;首次报道了pep20介导的CD47-SIRPα阻断可通过刺激IFN-γ分泌和下调系统Xc–来致敏铁凋亡。综上所述,该工作提出了一种新的联合模式来增强PDT,并揭示了新的联合机制,可能为乳腺癌的联合治疗提供新的途径。