背景
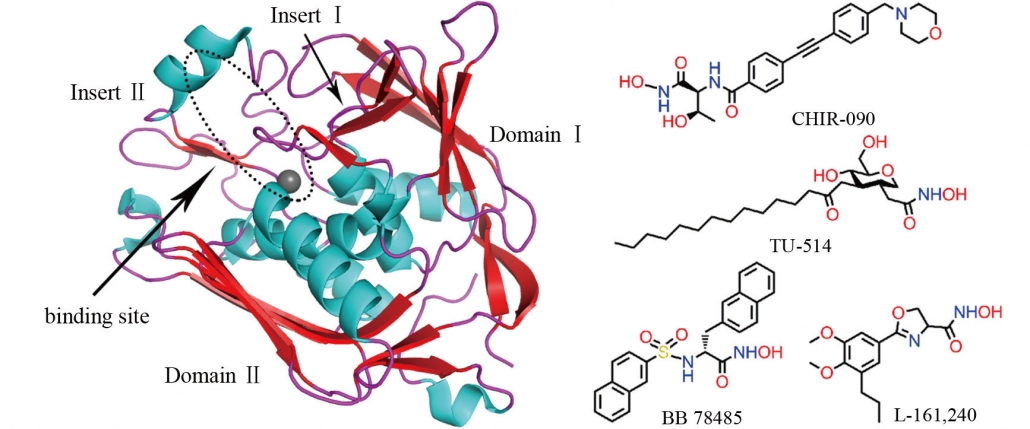
内毒素脂质A(lipid A,LA)作为G-菌脂多糖(lipopolysaccharide, LPS)嵌合于外膜上的“ 锚”, 其生物合成对于G-菌外膜组装至关重要。LA的合成共有9个酶参与, 其中由LpxC基因编码的UDP-3-O-(R-羟基十四酰)-N-乙酰氨基葡糖脱乙酰酶(LpxC)催化的脱乙酰反应是整个生物合成途径中的首个关键步骤,有效抑制LpxC能干扰LPS的合成,最终导致菌体细胞的死亡。另外, LpxC在G-菌中高度保守(超过40种),且在人和哺乳动物体内无同源性蛋白质。因此, LpxC是理想的抗菌药物新靶点,基于LpxC结构开发小分子Inhibitor或将改善耐药G-菌感染治疗困难的现状。
目的
通过同源模建(homology modeling)技术、分子对接(molecular docking)和分子动力学(molecular dynamics,MD)模拟等方法构建合理的AbLpxC结构,并从氢键和结合自由能等角度出发讨论典型LpxC Inhibitor与其的分子识别机制。
方法
同源模建
分子对接
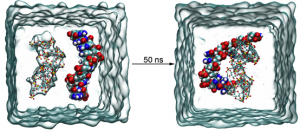
分子动力学模拟
主要研究结果
基于PaLpxC晶体结构, 通过同源模建技术模建了AbLpxC全酶模型, 并将最低PDF和DOPE值的构象作为最终模建结果。通过Ramachandran plot和Profile-3D参数验证了模型的可靠性。
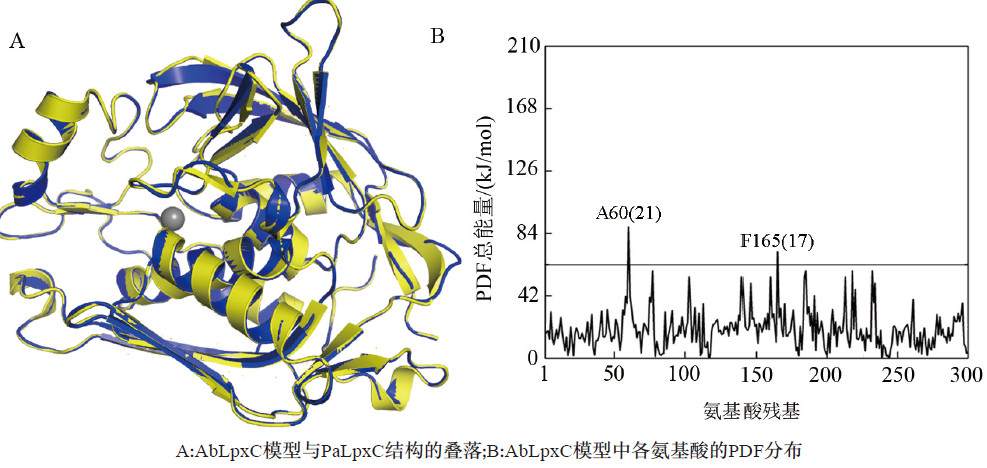
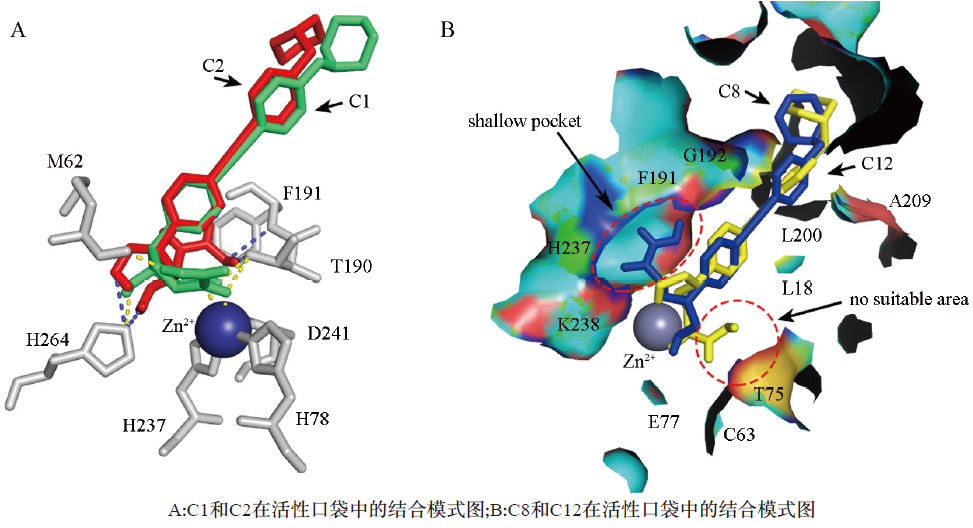
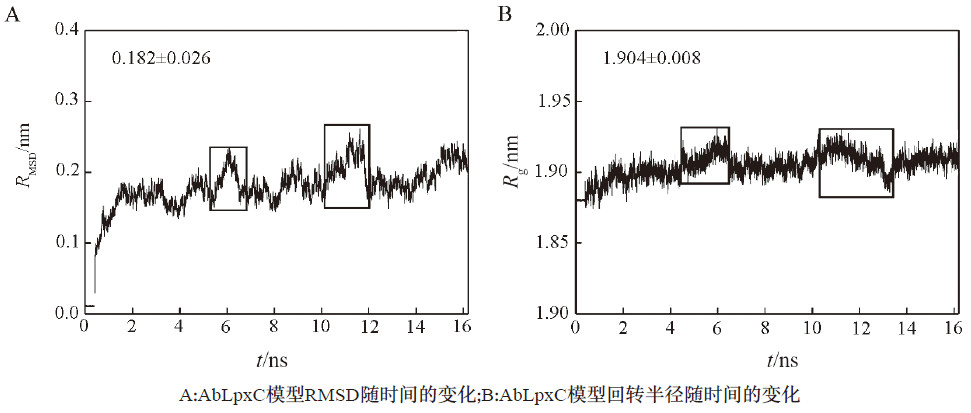
模建模型的97.3%位于最适区, 2.4%位于允许区, 仅A255残基位于禁止区。 随后采用MD模拟精修了AbLpxC模型, 势能、回转半径和RMSD值分析结果表明, 部分不合理的构象得到有效修正且整个模拟轨迹较为平稳。最后, 基于MD模拟平衡后能量最低构象与文献报道的12个BOAHAs抑制剂进行了分子对接,对接结果显示,C1为AbLpxC模型的最佳抑制剂, 其四氢呋喃环为影响分子识别的关键结构. 疏水链的长度对BOAHAs Inhibitor活性影响较大。
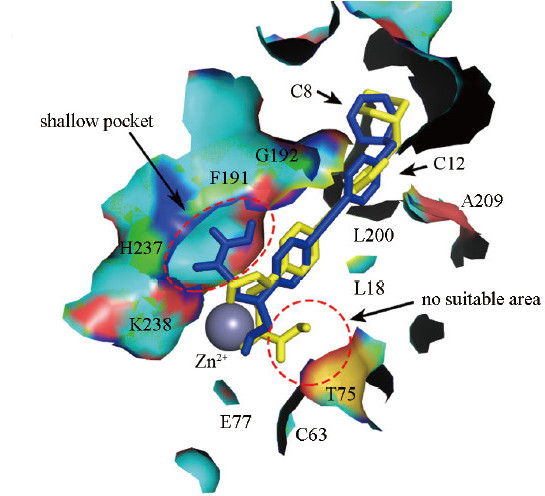
另外, 单手性结构中仅S构型能有效结合于F191,H237和K238组成的口袋,解释了R和S构型间结合自由能的差异,且与抑制活性实验测定结果吻合. 本文将为基于AbLpxC结构的新抗菌分子筛选、设计提供有用的结构信息。