合作单位:University of Chinese Academy of Sciences
参考文献:Yuan Zhuang, Xuechun Wang, Qiaozhi Liu, et al. Chemical Engineering Journal, 379, 122310-122310. DOI:10.1016/j.cej.2019.122310(IF=16.74)
本研究采用简单的一步水热法合成了N掺杂FeOOH/RGO水凝胶。用氨水控制α-FeOOH纳米盒的结晶生长,诱导双反应中心形成(Fe和C周围)和N掺杂在三维石墨烯基体中。所得水凝胶在较宽的pH范围内具有良好的稳定性和较强的四环素降解活性。
背景:
水污染是一个严重的全球性问题,开发能有效的去除水中抗生素的技术十分重要。高级氧化过程(AOPs)产生高活性自由基来降解有机污染物。铁的氧化物(包括FeOOH)由于对环境友好、数量丰富以及较强的•OH生成能力,是首选的非均相Fenton反应催化剂。然而,铁纳米粒子易聚集、难分离的特性限制了其应用。因此,可以开发适合的铁的氧化物载体,以提高铁的氧化物的活性和稳定性,解决分离问题。
石墨烯是一种纳米材料,由于其优异的热学、电学和机械性能被广泛的研究。抗生素一般含有π-电子和羟基,它们可以通过π-π相互作用、氢键和范德华相互作用被石墨烯很好地吸附。为了限制石墨烯片的重新堆积以获得活性位点并提高分离的便利性,将二维(2D)石墨烯集成到三维(3D)宏观水凝胶中已成为一种重要的方法。柔性石墨烯片可以在石墨烯水凝胶的三维空间内部分重叠,形成独特的具有多孔互联的层级结构,便于电子转移,这为金属催化剂创造了良好的载体。此外,氧化石墨烯(GO)片含有氧官能团,可以作为铁前体成核和生长的激活位点。石墨烯与FeOOH之间的Fe – O – C键是提高电子传递速率以促进Fe(III)/Fe(II)转化,提高非均相Fenton反应效率的重要环节。近年来,石墨烯/氧化铁基纳米复合材料因其有效降解药物的能力而受到了广泛关注。然而,可定向合成、形状可控的具有高比表面积及丰富的活性位点的石墨烯水凝胶的制备仍然是一个巨大的挑战。非均相Fenton催化剂通常需要中性条件来保证其催化活性,但Lyu等人通过在CuAlO2和氮化碳化合物之间建立Cu-O-C连接的双反应中心,创造了在宽pH范围内具有高污染物降解效率的催化剂,从而消除了这一限制。结果表明,金属与碳基材料在金属- O – C键中的相互作用是提高催化性能的有效途径。此外,通过加入N或Fe配位的N化合物可以提高低成本碳催化剂的催化活性。因此,我们有理由认为,在FeOOH/RGO水凝胶中建立强Fe – O – C键桥以建立双反应中心和N掺杂可以有效提高Fe(III)向Fe(II)的有限转化和非均相Fenton催化效率。
方案设计:
为了研究氧化石墨烯改性前后与四环素(TET)的结合模式以及二者结合的差异,经与魔德科技(www.modekeji.cn)技术团队沟通,本文采用分子动力学(Molecular dynamics, MD)模拟方法分别研究了TET在改性前后氧化石墨烯上的吸附情况。
主要结果:
- 分子动力学模拟整体构象变化
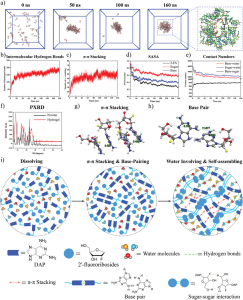
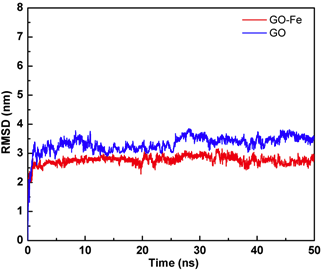
体系的均方根偏差(Root mean square deviation, RMSD)随时间的变化如图1所示,从图中可以看出,两个体系在模拟过程中已经趋于稳定,为后续基于分子动力学模拟的分析奠定了基础。
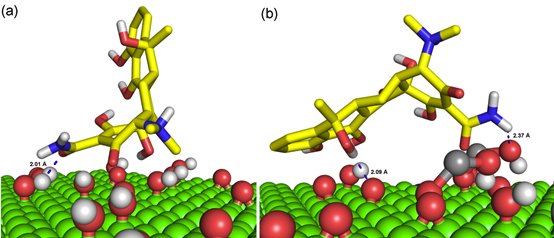
TET分子在GO和GO-Fe表面的吸附过程如图2所示,从图中可以看出,两个体系经过50 ns的分子动力学模拟,TET均能吸附到氧化石墨烯的表面。具体来讲,由于GO-Fe体系氧原子含量更高,能够提供的氢键供体/受体更多,因此其与TET的结合更加紧密。
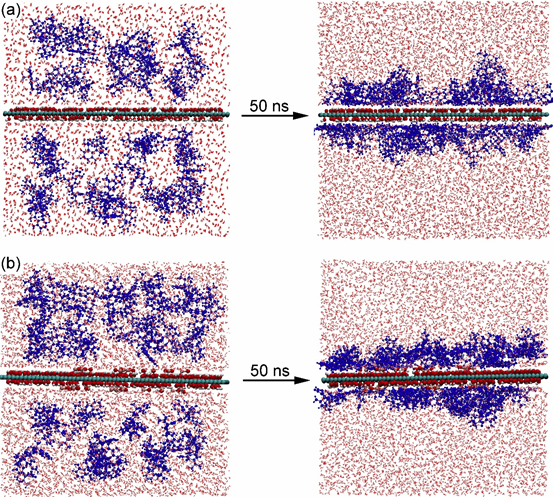
图3分别给出了两个体系中TET和水分子沿Z轴的密度分布。从图中可以看出,TET基本分布在石墨烯层的两侧,峰值出现在距离石墨烯层0.5 nm处,并且在GO-Fe体系(图5b)中,TET在峰值处的密度比GO体系(图3a)更大,表明TET在前者中的分布更加密集。
综上所述,通过分析两个体系在分子动力学模拟过程中的RMSD、密度以及构象变化,表明TET在GO-Fe表面的分布比GO表面更加紧密有序,因此推测TET与GO-Fe的亲和力更强。
- TET与GO/GO-Fe体系的相互作用
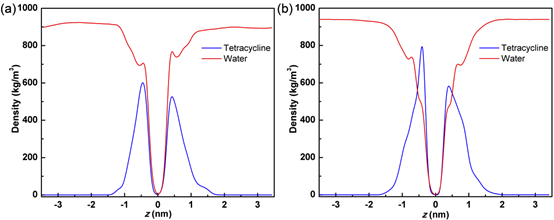
由于GO和GO-Fe表面都被氧原子修饰,而TET本身结构中也包含较多的氢键供体和受体,因此可以推测,氢键作用是TET与GO/GO-Fe结合的最重要的驱动力。图4给出了模拟过程中TET分子与GO和GO-Fe之间的氢键数目的变化。从图中可以看出,TET与GO-Fe之间的氢键数量明显比GO的更多,表明TET与GO-Fe之间的亲和力比后者更强。
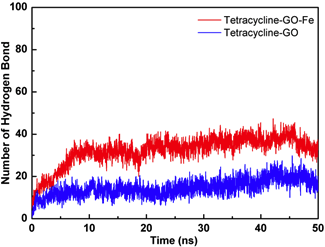
图5分别给出了TET在氧化石墨烯表面具有代表性的结合模式。由于GO体系中,石墨烯表面只有羟基或氧原子修饰,两种基团距离石墨烯平面都很近(≈2 Å),使得TET只能与GO形成较少的氢键作用。而在GO-Fe体系中,由于氧原子的含量更大并且有Fe原子的修饰,导致氧原子在石墨烯表面的分布更多,且能够达到距离石墨烯平面更远的位置(≈4 Å),TET也更容易与GO-Fe中的氧原子形成氢键作用,进而增强了二者的结合能力。
通过分析分子动力学模拟过程中的收敛参数的变化,发现TET在GO-Fe表面分布的密度比GO更大,并且结合更加稳定;通过分析模拟过程中的氢键数量以及代表性结合构象,发现由于GO-Fe中Fe原子的修饰以及氧原子的含量更大,使TET能够与其形成更多的氢键作用,进而TET与GO-Fe的结合能力更强。分子模拟结果可以在一定程度上解释氧化石墨烯及改性后的结构吸附四环素类抗生素的机制,并为后续研究提供理论指导。